

02.02.2018
2 minutes of reading




IFPEN développe une méthodologie visant à recaler des lois thermodynamiques empiriques, notamment présentes dans les simulateurs de bassin, sur des données générées in silico par une approche de simulation moléculaire.
La diminution des ressources d’hydrocarbures impose aujourd’hui l’exploitation de champs présentant toujours plus de difficultés techniques à surmonter, comme ceux contenant une forte concentration en hydrogène sulfuré (H2S). Ce gaz acide provient de la réaction de thermo-réduction des sulfates (TSR) dans le sous-sol et, outre sa forte toxicité, il peut corroder prématurément les infrastructures pétrolières qui se trouvent au contact du brut : par exemple des équipements de transport, de séparation ou de traitement. Aussi bien en phase d’exploration que de production, la présence d’H2S génère donc des risques et des coûts que les exploitants ont besoin d’anticiper.
La quantité d’H2S présente dans le sous-sol peut déjà être estimée à l’aide de logiciels de géoscience, tels que la suite OpenFlow™ et l’outil de modélisation de bassin TemisFlow™ lequel, pour la phase d’exploration, permet de simuler la génération et le transport de l’H2S issu de la réaction de TSR.
Cette modélisation, qui permet de déterminer la solubilité de l’hydrogène sulfuré dans des saumures (telles que rencontrées dans les gisements), repose sur des lois thermodynamiques qui sont calées au moyen de données expérimentales. Or, ce type de données est quasiment inexistant pour les conditions géologiques dans lesquelles se produit la réaction de TSR. Par ailleurs, compte tenu des propriétés de ce gaz acide, l’acquisition de nouvelles données en laboratoire présente un potentiel de dangerosité dont on souhaite s’affranchir.
C’est pourquoi IFPEN a élaboré une méthodologie qui vise à remplacer les expérimentations classiques. Elle consiste tout d’abord à générer des données « pseudo-expérimentales » de solubilité de l’H2S dans une saumure, des conditions opératoires d’intérêt, au moyen de calculs de simulation moléculaire (méthode Monte Carlo).
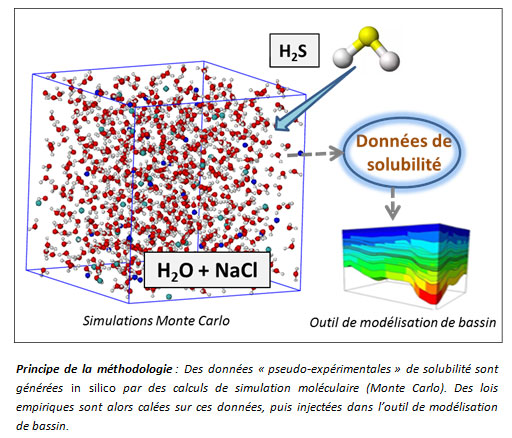
Par ses bases physiques rigoureuses, la simulation moléculaire permet de prédire avec une bonne précision les propriétés thermophysiques dans des conditions opératoires difficilement accessibles par voie expérimentale. Des lois thermodynamiques empiriques (équations d’état, modèles d’activité, etc.) sont alors recalées sur ces nouvelles données, avant d’être introduites dans le simulateur de bassin. Cette méthodologie s’apparente à de l’expérimentation in silico, et illustre pleinement les orientations d’IFPEN dans le domaine de la digitalisation.
Cette approche visant à utiliser la simulation moléculaire soulève l’intérêt des industriels pour d’autres applications des géosciences, notamment dans le domaine du stockage d’hydrogène au sein de formations géologiques.
Contacts scientifiques : Nicolas Ferrando - xavier.guichet@ifpen.fr - Véronique Lachet
Publication
- Fauve, R. ; Guichet, X. ; Lachet, V. ; Ferrando, N. Prediction of H2S solubility in aqueous NaCl solutions by molecular simulation. Journal of Petroleum Science and Engineering, 2017, 157, 94-106.
>> DOI: 10.1016/j.petrol.2017.07.003